Antarctic soil sample from 2018 adds to the enigma of the COVID-19 pandemic

The origin of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has long been debated among scientists. However, prior research found early genetic material of a unique SARS-CoV-2 variant from the soil on King George Island, Antarctica between 24 December 2018 and 13th January 2019. Following up on these findings, new research by scientists at the University of Veterinary Medicine, Budapest, suggests the new variant is related to genetic material from the mitochondria of Homo sapiens, green monkey, and Chinese hamster and likely came from cell lines Vero E6 in green monkeys and Chinese hamster ovary cells, which is frequently used to study coronaviruses in the lab.
 Study: Host genomes for the unique SARS-CoV-2 variant leaked into Antarctic soil metagenomic sequencing data. Image Credit: Alex.Munoz / Shutterstock
Study: Host genomes for the unique SARS-CoV-2 variant leaked into Antarctic soil metagenomic sequencing data. Image Credit: Alex.Munoz / Shutterstock
“The absence of more precise information on the contaminator samples leaves open the possibilities that either there are known samples that have escaped our attention, or that there are unpublished results that may be key to identify the origin of SARS-CoV-2,” concluded the research team. However, the researchers caution that these findings are preliminary, and have not been confirmed as a scientific fact.
The study “Host genomes for the unique SARS-CoV-2 variant leaked into Antarctic soil metagenomic sequencing data” was recently posted to the Research Square preprint* server. For clarity, a research preprint is a preliminary version of a manuscript that has not completed the peer review process at a journal.
Study background
The Antarctic soil samples containing SARS-CoV-2 sequence fragments were sent to a lab in Shanghai, China, in December 2019. A phylogenetic analysis of the samples suggests they are early variants related to the Pango lineage “A” characterized by rare mutations and a long deletion at nucleotide 21761.
The researchers of the current study do not believe that SARS-CoV-2 originated in Antarctica but instead were mistakenly assigned reads from another SARS-Cov-2 sample sequenced on the same flow cell at the Sangon Biotech sequencing facility. This is likely because the equipment used to sequence the data is prone to index hopping and misassignment errors from contaminated paired-end samples with only the R2 mates of SARS-CoV-2.
The current study looks into finding the genetic footprint of the hosts harboring the discovered viruses.
Study details
The researchers hypothesized that the host or hosts’ genetic material may have leaked into viral sequencing data as well. Because the size of a mammalian mitochondrial genome is similar to a coronavirus, the likelihood of high genome coverage in a single mammalian cell is high because they have thousands of mitochondria with their own genome.
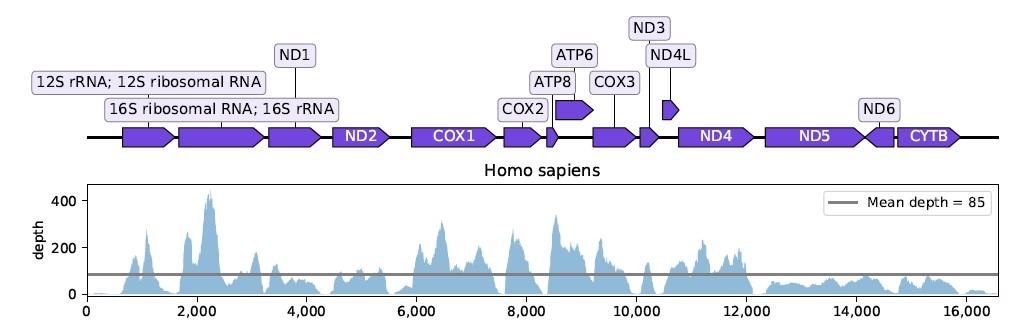
The smoothed coverage of the human mitochondrial reference genome by the union of R2 reads from samples SRR13441704, SRR13441705 and SRR13441708. The average depth is 85 but there are large fluctuations in the coverage.
The researchers used the current database containing information of all types of mitochondria to narrow their search into mammals only. Using the term “Vertebrata” in their taxonomy, the team landed 6,158 possible species. They then compared the vertebrate mtDNA reference genome set to the reads from viral genome samples.
To avoid false or misleading hits, the researchers calculated the percentage of nucleotides covered in the mitochondrial genome and the average number of reads in a nucleotide (average depth) for each species
Results showed that samples RR13441704, SRR13441705, and SRR13441708 were the most abundant in SARS-CoV-2 reads compared to two other samples where researchers could not find SARS-CoV-2 reads. Samples with SARS-CoV-2 reads had more extensive mtDNA content.
Additionally, for certain species, the samples with SARS-CoV-2 reads had orders of magnitudes higher coverage than samples that did not have SARS-CoV-2 reads. The extra accounts came from the Homo sapiens, Cricetulus griseus (Chinese hamster), and the Chlorocebus sabeu (green monkey) species.
Because the large number of mtDNA correlated with the abundance of the SARS-CoV-2 content, the mtDNA reads likely came from the same contaminor samples. Further, the researchers explain this is considerable evidence to show Homo sapiens, Cricetulus griseus, and the Chlorocebus sabeu species are hosts of the detected SARS-CoV-2.
Because none of the mammals live in the Artic, the findings suggest SARS-CoV-2 did not come from Antarctica but rather contaminated in samples of their cells.
The most probable scenario is that they were contaminated cells in a lab as the Chinese hamster is used to study other betacoronaviruses such as MERS-CoV and SARS-CoV. The Vero cell line in green monkeys is used to study the Marburg virus.
*Important Notice
Research Square publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Csabai I & Solymosi N. (2022). Host genomes for the unique SARS-CoV-2 variant leaked into Antarctic soil metagenomic sequencing data. Research Square. Doi: 10.21203/rs.3.rs-1330800/v1, https://www.researchsquare.com/article/rs-1330800/v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Cell, Cell Line, CLARITY, Coronavirus, Coronavirus Disease COVID-19, Genetic, Genome, Marburg Virus, Medicine, MERS-CoV, Mitochondria, Nucleotide, Nucleotides, Pandemic, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Veterinary, Virus

Written by
Jocelyn Solis-Moreira
Jocelyn Solis-Moreira graduated with a Bachelor's in Integrative Neuroscience, where she then pursued graduate research looking at the long-term effects of adolescent binge drinking on the brain's neurochemistry in adulthood.
Source: Read Full Article