nifedipine ir
<img class="aligncenter" src="https://scx1.b-cdn.net/csz/news/800a/2023/treating-a-heart-attac.jpg"
alt="Treating a heart attack before it happens"
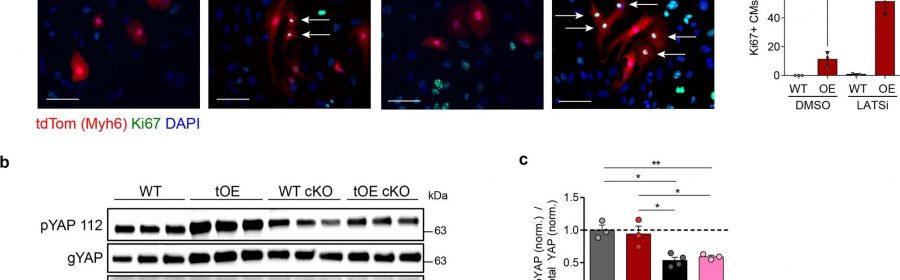
title="LATS1/2 negative feedback signaling is required for redifferentiation. a, Representative immunofluorescence images of Ki67 in LATSi or DMSO treated WT and OE P7 cardiac cultures, with full quantification. All groups had minimum n = 3. Scale bars = 50μm. b,c, Representative western blot of whole-heart lysates for general Yap (gYAP) and pYAP S112 (a target residue of LATS1/2) from WT, tOE, how can i get promethazine codeine from my doctor WT LATS1/2 cKO and tOE LATS1/2 cKO mice (b), with quantification, normalized to the average WT value (c). WT n = 3, tOE n = 3, WT cKO n = 4, OE cKO n = 3. In all panels numerical data are presented as mean ± SEM; statistical significance was calculated using a paired two-way ANOVA followed by Sidak’s test in (a) and a one-way ANOVA followed by Tukey’s test in (c). *p ≤ 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Uncropped blots for (b) are provided in supplementary source data. Credit: Nature Cardiovascular Research (2023). DOI: 10.1038/s44161-023-00250-w” width=”800″ height=”351″>
Imagine getting treatment for a perfectly healthy young heart that would allow it to recover from an otherwise devastating injury decades later.
If you think this prospect seems farfetched, you are not alone. Until recently, Prof. Eldad Tzahor, whose lab at the Weizmann Institute of Science studies heart tissue regeneration, had also considered it science fiction. After all, cardiovascular diseases, which are humanity’s leading cause of death, aren’t generally perceived as something one can prepare for through preventive treatment.
But Tzahor and researchers in his lab have now activated a cellular mechanism in healthy mouse hearts that makes these mice resilient to future heart attacks—even when they occur months later.
The procedure is a long, long way from being applicable to humans, the researchers stress. But their findings, published today in Nature Cardiovascular Research, reshape our understanding of the regenerative capabilities of the heart—and possibly other organs—and how they might be enhanced through preventive medical intervention.
“It’s a proof of concept,” says Tzahor, “and it points to new avenues of research that examine giving heart treatment not only after the damage happens, but from a preventive position that increases the capacity for recovery from an injury before the damage even occurs.”
The study, led by Dr. Avraham Shakked in Tzahor’s lab in Weizmann’s Molecular Cell Biology Department, focused on genetically engineered mice whose cardiomyocytes—the cells that make up the heart muscle tissue—overexpress a gene that triggers cell division in mice and other mammals, including humans. In previous studies scientists in Tzahor’s lab had found that the gene, ERBB2, causes cell division in cardiomyocytes—a remarkable occurrence because at around the time of birth these cells lose their capacity to multiply.
“During fetal development, our cells are assigned their different roles—nerves, cornea, heart muscle etc.—through a process called differentiation,” says Shakked. “It’s characterized by a spectrum: On one end are stem cells, which are undifferentiated but capable of dividing and producing various types of cells. On the other end are highly-specialized cells like cardiomyocytes, which are no longer able to divide after becoming differentiated. They’re very effective in their function, but the tissue they comprise does not regenerate naturally.”
This is one of the reasons that cardiac episodes are so devastating. Heart attacks kill off massive numbers of cardiomyocytes that the body cannot regenerate. Therefore, even individuals who survive the attack are often left with reduced cardiac performance.
When, in previous studies, Tzahor’s team had managed to trigger the division of cardiomyocytes—by switching on ERBB2 briefly in these cells—overall heart function actually decreased temporarily, rather than improving straightaway. This happened because the ERBB2-expressing cardiomyocytes underwent dedifferentiation, meaning that they reverted to a less specialized state, closer to that of a fetal heart. This, in turn, limited their ability to contract, which is needed for proper heart function.
But once the overexpression had stopped, the cardiomyocytes underwent redifferentiation—that is, they became highly specialized again—and cardiac performance improved.
In the new study, the scientists sought to understand what happens to hearts “rejuvenated” by ERBB2 and how, exactly, they redifferentiated and returned to normal function once the gene had been switched off. The performance of such hearts was indistinguishable from that of the control group, but Shakked did notice some significant differences in gene expression between the two populations. “It was surprising and curious,” he recalls.
“We had assumed that everything returns to normal after ERBB2 is switched off in the cardiomyocytes. Yet here we were, seeing a different genetic pattern—overexpression in some genes and underexpression in others—following ERBB2 activation. In other words, we found long-term effects.”
This discovery made Shakked and Tzahor wonder whether ERBB2 expression could be calibrated for improved cardiac performance. “It made us think that ERBB2 wasn’t just a switch that prevents differentiation, but part of a mechanism that could make the heart younger and more resilient,” Tzahor says.
To test this hypothesis, the researchers reversed the order of their previous experiments with ERBB2. Instead of switching ERBB2 on in injured mice to make their cardiomyocytes divide, they first switched it on in healthy mice for a few weeks and then switched it off again. Next, the researchers observed how the hearts of those mice coped with an injury.
The result: Mice that had been made to overexpress ERBB2 recovered, but others did not. “The data made our jaws drop,” Tzahor recalls. “We had found a cardiac fountain of youth in those mice, a novel way of making the heart younger and stronger.”
Currently, the research team is examining a number of hypotheses about mechanisms through which a brief overexpression of ERBB2 could help mice survive future heart damage. One possibility is that the gene triggers a series of changes that allow more cardiomyocytes to survive the lack of oxygen that is characteristic of heart attacks and is particularly destructive for cardiomyocytes.
The researchers also revealed how a negative feedback loop drives redifferentiation in cardiomyocytes. “The body makes sure that differentiation happens because, generally, it doesn’t want cells that do nothing but divide, like cancer cells,” Tzahor says.
“That’s why ERBB2, while moving cardiomyocytes in the opposite direction of natural differentiation, simultaneously activates genes that trigger differentiation. There are checks and balances in place. If it weren’t for this mechanism, the dedifferentiated heart cells would not have been able to redifferentiate back to functioning cardiomyocytes,” Tzahor explains.
The team found that a mouse whose ERBB2 had been temporarily activated when it was three months old recovered from major cardiac injury that happened five months later. “If we translate this to human years, it’s comparable to an 18-year-old getting treatment that allows that person to survive a heart attack at age 50,” Tzahor says.
However, this kind of treatment is at present nowhere near being applicable to human beings. “We’re lowering the function of cardiomyocytes to allow them to be restored in the future,” Tzahor explains. “From a clinical perspective, this is an extreme and drastic intervention. Still, at least in principle, our research might lead to a way of treating people who have a high risk of heart attack, before these attacks even happen.”
More information:
Avraham Shakked et al, Redifferentiated cardiomyocytes retain residual dedifferentiation signatures and are protected against ischemic injury, Nature Cardiovascular Research (2023). DOI: 10.1038/s44161-023-00250-w
Journal information:
Nature Cardiovascular Research
Source: Read Full Article