Scientists uncover key role for proteins involved in amyotrophic lateral sclerosis disease

 and depletion of OPTN delay clearance of SGs and increases ubiquitinated TDP-43 levels by increasing TIA1. Credit: Niigata University")
Stephen Hawking, the world-famous theoretical physicist, was diagnosed with amyotrophic lateral sclerosis (ALS) at the age of 21. ALS is a rare motor neuron disease that usually affects individuals over the age of 60. Hawking survived for another 55 years, defying the odds. Hawking’s illness, on the other hand, is an anomaly, since the majority of patients live just around 2-4 years after diagnosis. While the disease is rare, it has a major socioeconomic impact since people suffer motor degeneration and, ultimately, paralysis.
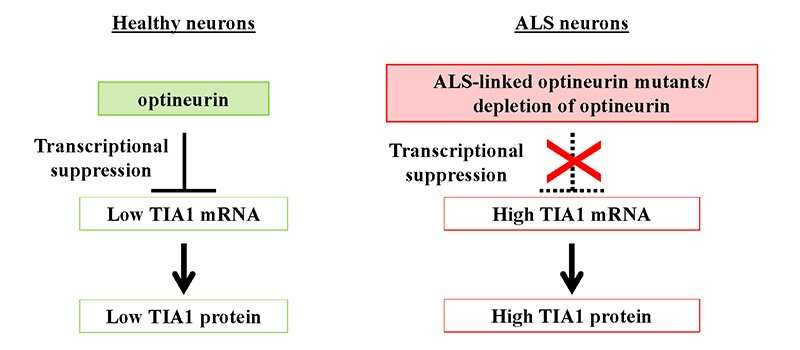
Although the cause of ALS is unclear, a team of Japanese scientists headed by Prof. Masahiro Fujii of Niigata University Graduate School of Medical and Dental Sciences and the Brain Research Institute is making significant progress in unraveling the underlying causative mechanisms. The accumulation of ubiquitinated TDP-43 (Ub-TDP-43) protein aggregates in motor neurons is a hallmark of ALS, and Prof. Fujii and colleagues have shown that a protein called optineurin (OPTN) inhibits the formation of Ub-TDP-43 aggregates in stress-induced stress granules (SGs), whereas the OPTN mutants derived from the ALS patients induce Ub-TDP-43 aggregates in SGs.
These SGs are generated in the cytoplasm of cells in response to a number of stressors, including heat shock and oxidation, and are subsequently eliminated spontaneously when the degree of stress diminishes. In their recent work, Prof. Fujii and fellow scientists, produced OPTN knockdown (OPTN-KD) cells and found an increase in the size of SGs, a delay in SG clearance and an accumulated Ub-TDP-43 in SGs after stress removal, which is characteristic of aberrant SGs. Additionally, they established a connection between this and an increase in the expression of TIA1, another protein implicated in the development of ALS.
This team of scientists demonstrated a direct link between OPTN-KD-induced increases in TIA1 protein and the formation of Ub-TDP-43-positive SGs in OPTN-KD cells.
Due to the delayed clearance of these aberrant SGs, neurons accumulate Ub-TDP-43 aggregates, which show toxicity to neurons, culminating in the characteristic neuron degeneration seen in ALS patients.
“This discovery elucidates the process by which Ub-TDP-43 aggregates develop in the brains of ALS patients with optineurin or TIA1 mutations and explains how ubiquitinated proteins aggregate in neurodegenerative disorders such as ALS,” Prof. Fujii explained in an interview.
Genetic variables, particularly mutations, are also important in ALS, particularly familial ALS. Previously, mutations in the OPTN and TIA1 genes were identified as causative factors in familial ALS with TDP-43 aggregation pathology. “Our findings indicate that ALS-associated OPTN loss-of-function mutations increase the quantity of Ub-TDP-43 in neurons via raising the expression of TIA1, thus enhancing Ub-TDP-43 aggregation,” added Prof. Fujii.
To further understand the mechanisms by which OPTN suppresses TIA1 expression, the scientists looked at the NF-κB signaling pathway, since previous research has shown that OPTN inhibits NF-κB-mediated gene transcription. On the other side, the OPTN-KD increased the levels of TIA1 mRNA, whereas an NF-κB inhibitor had no effect, suggesting that this process is not NF-κB dependent. While it is a breakthrough that this study establishes a significant link between OPTN and aberrant SG formation, elevated TIA1 expression, ubiquitination, and TDP-43 aggregation, future research efforts should be directed toward understanding the mechanism by which OPTN regulates TIA1.
Source: Read Full Article