Mapping the trajectory of SARS-CoV-2 evolution inside the host

In a recent study posted to the bioRxiv* pre-print server, researchers in the United States mapped the trajectory of within-host evolution of acute severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection.
 Study: Within-host evolutionary dynamics and tissue compartmentalization during acute SARS-CoV-2 infection. Image Credit: ktsdesign / Shutterstock
Study: Within-host evolutionary dynamics and tissue compartmentalization during acute SARS-CoV-2 infection. Image Credit: ktsdesign / Shutterstock
Background
The large-scale whole genomic sequencing efforts on a global scale and phylogenetic analyses of clinical samples during the coronavirus disease 2019 (COVID-19) pandemic have captured the global evolutionary dynamics of SARS-CoV-2. However, there is a lack of understanding of SARS-CoV-2 evolutionary dynamics inside the host.
Although a few studies have previously captured within-host SARS-CoV-2 dynamics but focused only on immunocompetent hosts. These studies showed low within-host diversity, with most samples containing 15 or fewer intra-host single-nucleotide variants (iSNVs). Together, data from these studies demonstrated that the selection-driven emergence of iSNVs to high frequency during acute infection is likely rare. Overall, a high-resolution profile of within-host SARS-CoV-2 evolutionary dynamics is missing.
Furthermore, it remains poorly understood how pre-existing immunity, elicited through vaccination or prior infection, influences the within-host evolution of SARS-CoV-2. More importantly, characterizing the potential for the emergence of immunity escaping variants in immune-competent individuals with different vaccination statuses is needed.
About the study
In the present study, researchers recruited 32 students, faculty, and staff members at the University of Illinois in the United States (US) for longitudinal sampling that allowed high-confidence SARS-CoV-2 variant detection to uncover evolutionary dynamics overlooked by less-frequent sampling strategies. Of these 32 study participants, 20 were naive individuals, and 12 had pre-existing immunity acquired via vaccination or natural SARS-CoV-2 infection. From each individual, the team collected mid-turbinate (MT) nasal swabs and saliva from both naïve and immune individuals daily for repeated measures of iSNV frequencies during the early infection phase. In this way, they generated high-resolution profiles of iSNV dynamics between tissue compartments and temporally.
Study findings
The indications of strong positive selection were rare in the study cohort. However, the researchers noted quite a few non-synonymous substitutions, including N: P67S, S: Q677H, and ORF1ab: P5402H from below detection limit to high frequency. Substitution at S: Q677 independently emerged in multiple SARS-CoV-2 sub-lineages around the world, supporting that mutations at this site can have an evolutionary advantage. The S: Q677H frequency was 56.5% when the associated study participant had a detectable infectious viral load in a nasal swab, indicating the forward transmission potential of this iSNV.
Further, the authors observed competition between S: Q677H and S: P681H substitutions within the same individual, with S: Q677H emerging to a high frequency for some time on a day at which the initially fixed frequency of S: P681H declined. However, the observed reversion to an S: P681H-only genotype after day 7 showed that the selective advantage conferred by S: P681H was greater than that of S: Q677H. The widespread proliferation of S: P681H-containing SARS-CoV-2 lineages in comparison to S: Q677H further supports the fitness advantage conferred by this mutation.
Further, genome mapping revealed the accumulation of multiple hotspots of non-synonymous mutations differing between naïve and immune individuals. The observed enrichment of amino acid substitutions immediately upstream of the SARS-CoV-2 spike subunit 1 (S1)/S2 cleavage site indicated that this region may be subject to stronger within-host selection in humans. Therefore, in naïve individuals, they identified hotspots at residues 402-457 in ORF1ab, and 655-681 in S, directly adjacent to the S1/S2 cleavage site. S1/S2 cleavage site substitutions are characteristic features of the Omicron, Delta, and Alpha SARS-CoV-2 lineages. A recent study by Y. Liu et al. demonstrated that substitutions at S1/S2 cleavage site have been responsible for the Delta variant’s increased relative fitness compared with Alpha.
A high density of nucleocapsid (N) gene substitutions in immune study participants further validated previous data suggesting the significance of the N gene during human adaptation. Accordingly, the researchers observed a hotspot of mutation accumulation in N:199-204.
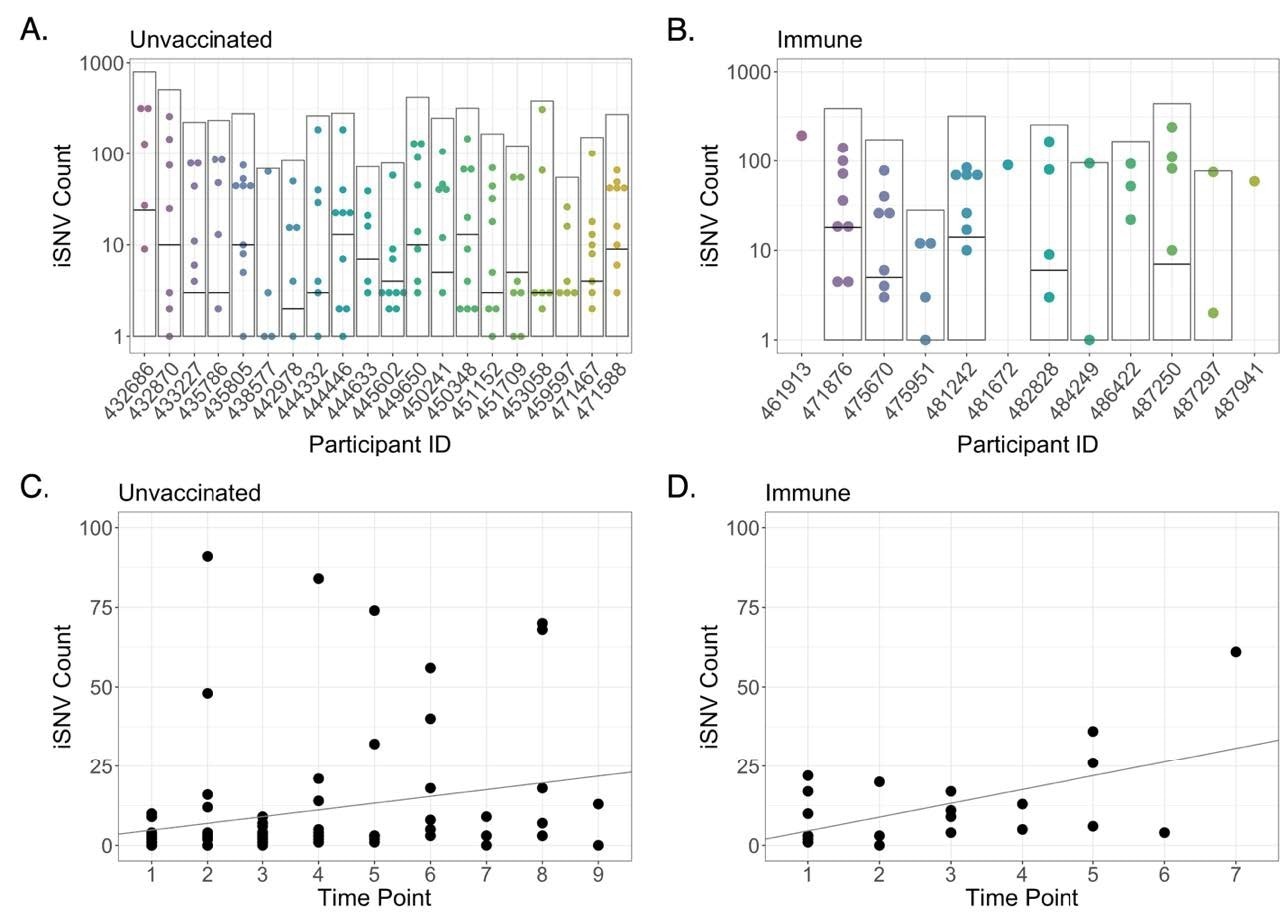
Intra-host single nucleotide variant (iSNV) diversity compared between samples and individuals. (A) Total iSNV counts for each sample from each unvaccinated participant. Light grey boxes indicate total iSNV count for all samples and horizontal black lines indicate number of shared iSNVs for each participant. (B) iSNV counts for immune participants. (C) iSNV counts for individual samples with Ct < 25 from naïve participants as a function of number of days post-enrollment (Adjusted R-squared = 0.05007, p = 0.02255). Line represents linear regression. (D) iSNV counts for individual samples with Ct < 25 from immune participants as a function of number of days post-enrollment (Adjusted R-squared = 0.2857, p = 0.006359). Line represents linear regression.
The researchers also observed several shared mutations within untranslated regions of the SARS-CoV-2 genome, such as the three prime untranslated region (3’ UTR). The most frequent was a t29760c substitution in the 3’ UTR, shared across nine naïve individuals. Future studies should investigate whether the recurring UTR mutations observed in the current study affect within-host SARS-CoV-2 fitness.
Furthermore, multiple study participants had extreme fluctuations at or near iSNVs. They abruptly fell below the detection limit and returned to high frequencies days later. It was likely due to spatial structuring, as has been described for the influenza virus by Amato et al. 2021. Spatial structuring promotes drift-driven fluctuations in sampled iSNVs due to bottleneck effects potentially arising from a poor quality sampling of the viral population. This finding further emphasizes the advantages of longitudinal sampling.
Finally, the researchers observed significant tissue compartmentalization between the oral and nasal environments throughout SARS-CoV-2 infection in some study participants. This explains why sampling of a single tissue site may not provide a complete picture of SARS-CoV-2 diversity within a host.
Conclusions
The study data provided a high-resolution profile of within-host SARS-CoV-2 evolutionary dynamics. The study results showed that within-host SARS-CoV-2 evolution in both naïve and immune individuals appeared primarily driven by stochastic forces during acute infections. In addition, the researchers identify mutational hotspots within the SARS-CoV-2 genome, consistent with the selection pressure that promotes the emergence of iSNVs capable of onward transmission.
Further, they also detected significant tissue compartmentalization of SARS-CoV-2 between nasal swabs and saliva samples in many individuals. Furthermore, recurrent detection of both successful and not-so-successful iSNVs at the global scale indicated areas of alliance and discordance between within-host and between-host selective pressures. This data shed light on the forces shaping the global patterns of SARS-CoV-2 evolution.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Within-host evolutionary dynamics and tissue compartmentalization during acute SARS-CoV-2 infection, Mireille Farjo, Katia Koelle, Michael A. Martin, Laura L Gibson, Kimberly KO Walden, Gloria Rendon, Christopher J. Fields, Fadi Alnaji, Nicholas Gallagher, Chun Huai Luo, Heba H. Mostafa, Yukari C Manabe, Andrew Pekosz, Rebecca Lee Smith, David D McManus, Christopher B Brooke, bioRxiv pre-print 2022, DOI: https://doi.org/10.1101/2022.06.21.497047, https://www.biorxiv.org/content/10.1101/2022.06.21.497047v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Coronavirus, Coronavirus Disease COVID-19, covid-19, CT, Evolution, Frequency, Gene, Genome, Genomic, Genomic Sequencing, immunity, Influenza, Mutation, Nucleotide, Omicron, Pandemic, Proliferation, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, students, Syndrome, Virus

Written by
Neha Mathur
Neha is a digital marketing professional based in Gurugram, India. She has a Master’s degree from the University of Rajasthan with a specialization in Biotechnology in 2008. She has experience in pre-clinical research as part of her research project in The Department of Toxicology at the prestigious Central Drug Research Institute (CDRI), Lucknow, India. She also holds a certification in C++ programming.
Source: Read Full Article