Investigating taraxerol as a protease inhibitor against SARS-CoV-2

In a recent study posted to the Research Square preprint* server, a team of researchers investigated the toxicity and absorption, distribution, metabolism, and excretion (ADME) characteristics of a potent herbal drug, taraxerol, in the management of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. The paper is under consideration for publication in the Structural Chemistry journal.
The coronavirus disease 2019 (COVID-19) pandemic has massively impacted human lives, with over 397 million confirmed cases and more than 5.74 million deaths worldwide to date. The search for a therapeutic medicine or preventive treatment against SARS-CoV-2 has led to the development of viral protease enzyme inhibitors.
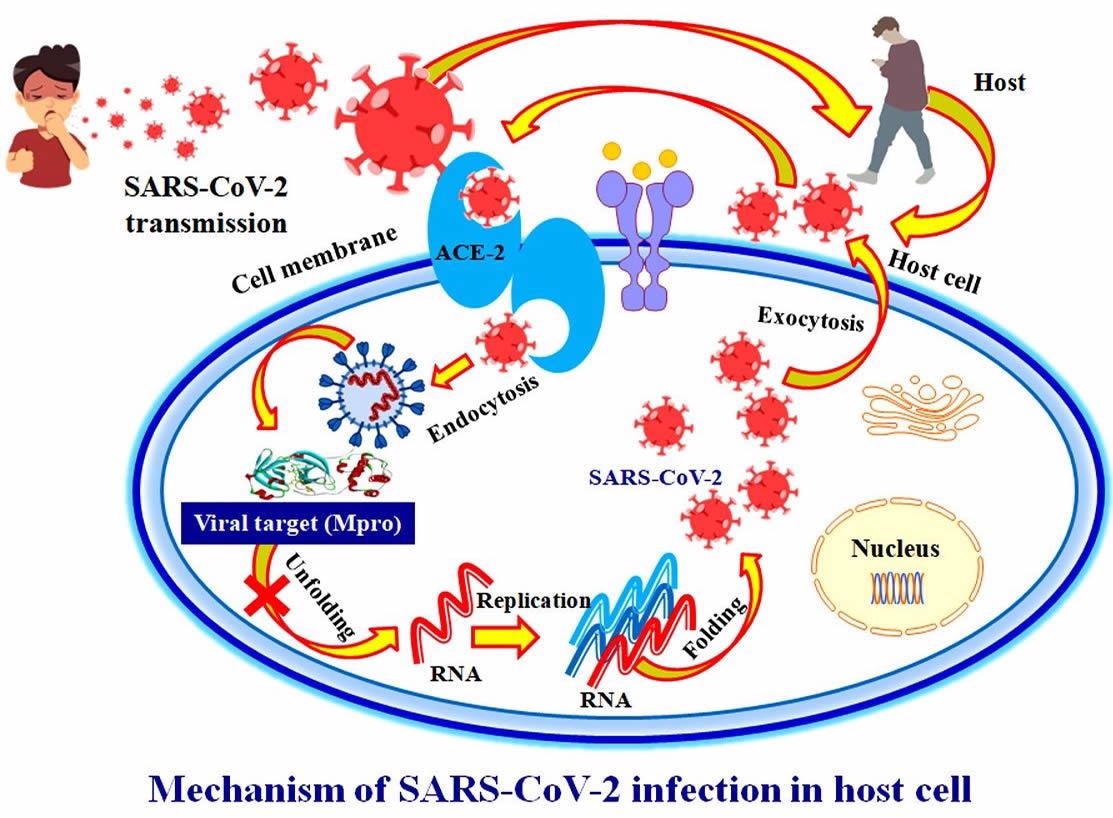
An in silico approach for screening herbal leads for potential inhibitors of the viral main protease enzyme in order to discover new antiviral therapies against SARS-CoV-2.
About the study
The present in-silico screening bioprospecting study examined the potential of taraxerol, a naturally-occurring pentacyclic triterpenoid, in managing SARS-CoV-2 infection. To put it simply, in silico refers to biological experiments carried out using computers or computer simulations and is a reliable, prudent, and quick method for identifying potential herbal compounds that are active against SARS-CoV-2's main protease enzyme.
The research involved the docking of the SARS-CoV-2 protease enzyme by amputating superfluous water molecules and adding polar hydrogens.
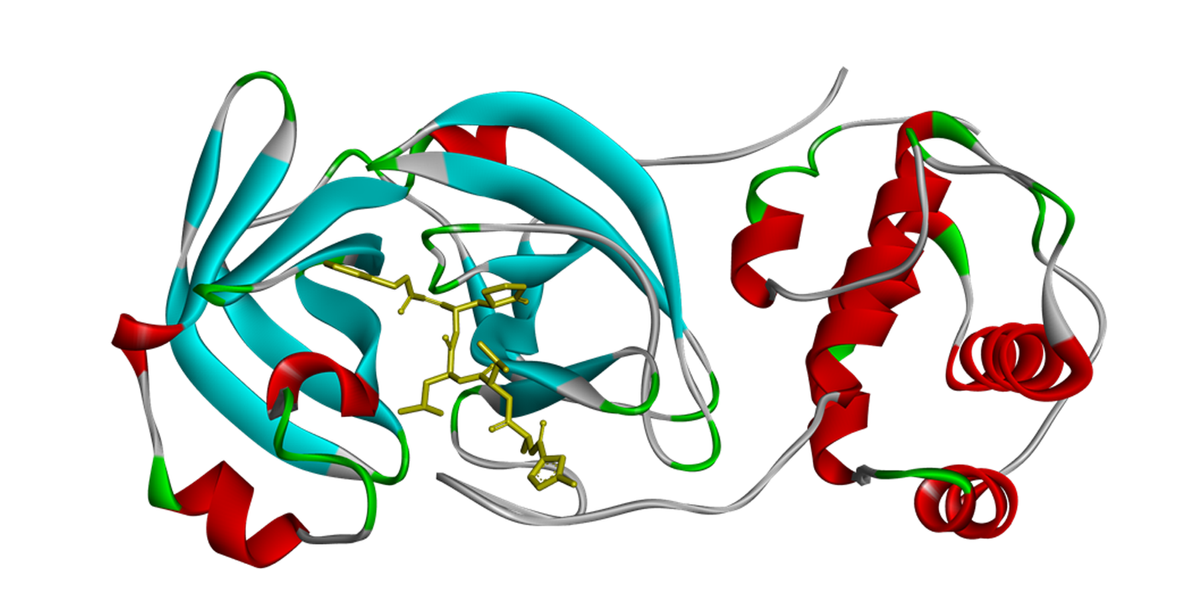
Three-dimensional structural model of the main protease enzyme complex with an antagonist.
A reference ligand was assembled by assigning un-rotatable, non-rotatable, and rotatable bonds. The active site of the protease enzyme was confirmed by examining the interaction of the viral molecular enzyme with the complex inhibitor. The chemical configuration of the viral molecule and the ligand was examined to conduct atomic mapping to simulate the docking process. Docking was executed to calculate the binding energy of the ligand by integrating the intermolecular energies and assessing energies of bound and unbound states.
A total of 150 herbal ligands were used to compare the ligands with the viral main protease enzyme via in silico screening. Furthermore, the ligands were virtually screened and compared to the viral protease enzyme. The outcomes of the docking process were evaluated using ligand-macromolecule interactions. Compounds with a binding energy between -5 kcal/mol and -15 kcal/mol were chosen as lead molecules.
The selected compounds were then screened and the macromolecular conformation stability with respect to time was evaluated using their docking score, non-involvement in other physiological processes, and safety profile. Molecular dynamics simulation for a duration of 10 nanoseconds (ns) followed by confirmation with magnified simulations lasting 100 ns was performed for the selected compounds. The macromolecular complex was solvated to perform the dynamic simulations. Electrostatic ligand-macromolecule interactions were calculated and the interactions were used to evaluate the binding interactions of the ligand and the protease enzyme of the macromolecule.
The study compared the reference ligand to the ligand-macromolecule complex to determine the atomic displacement and calculate the root mean square deviation (RMSD). Root mean square fluctuation (RMSF) was used to calculate the initial condition of the macromolecule in the crystalline structure. Also, the macromolecular secondary structural elements (SSE) were identified as alpha-helices, beta-strands, etc.
Results
The study results showed that the viral protease enzyme constituted a single chain of a total of 306 amino acid residues. The affinity of the molecules towards the protease was used as the selection criteria for lead molecules. The binding affinity of the lead molecules was determined by evaluating their interactions with the macromolecule. Among the selected lead molecules, diosgenin, amyrin, and taraxerol were chosen to study the stability of the conformation of the macromolecular complex. Taraxerol was the most stable lead compound against the protease enzyme, achieving the highest stability inside the binding site of the protease.
The RMSD value of the macromolecular residues was 0.88, which indicated the ligand-macromolecule complex did not cause any positional shift in most of the residues. The SSE analysis showed the presence of 14.94% and 24.21% alpha helices and beta sheets, respectively, with a total of 39.14% SSE conserved through almost the entire simulation phase. Consistent interactions of over eight macromolecular residues with the complex ligand were observed.
The binding energy sufficient for the inhibition of protease enzyme was observed in lead molecules with taraxerol at -10.17 Kcal/mol, diosgenin at -10.12 Kcal/mol, amyrin at -9.56 Kcal/mol, asiaticoside at -9.54 Kcal/mol, momordicin at -9.51 Kcal/mol, hecogenin at -9.42 Kcal/mol, guggulsterone at -9.23 Kcal/mol, andrographolide at -8.61 Kcal/mol, pelargonidin at -8.49 Kcal/mol, and lupeol at -8.48 Kcal/mol.
Conclusion
These bioprospecting study findings presented the therapeutic potential of herbal drugs like taraxerol against SARS-CoV-2 infections by inhibiting the viral protease enzyme. Amino acid residues were observed to play an essential role in the ligand-macromolecular interaction and binding.
The docking simulation and the pharmacokinetic profiling proved the efficiency of taraxerol as a viral protease enzyme inhibitor in therapeutic use. However, preclinical and clinical studies of taraxerol against COVID-19 are necessary to validate its clinical efficacy and safety further.
*Important notice
Preprints with Research Square publish preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Somdutt Mujwar, Ranjit K. Harwansh. In-silico bioprospecting of taraxerol as a main protease inhibitor of SARS-CoV-2 to develop therapy against COVID-19, 03 February 2022, PREPRINT (Version 1) available at Research Square, DOI: https://doi.org/10.21203/rs.3.rs-1308726/v1, https://www.researchsquare.com/article/rs-1308726/v1
Posted in: Medical Research News | Disease/Infection News
Tags: Amino Acid, binding affinity, Compound, Coronavirus, Coronavirus Disease COVID-19, covid-19, Drugs, Efficacy, Enzyme, Ligand, Medicine, Metabolism, Molecule, Pandemic, Preclinical, Research, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome

Written by
Susha Cheriyedath
Susha has a Bachelor of Science (B.Sc.) degree in Chemistry and Master of Science (M.Sc) degree in Biochemistry from the University of Calicut, India. She always had a keen interest in medical and health science. As part of her masters degree, she specialized in Biochemistry, with an emphasis on Microbiology, Physiology, Biotechnology, and Nutrition. In her spare time, she loves to cook up a storm in the kitchen with her super-messy baking experiments.
Source: Read Full Article